
Independent consultancy NNE and venture advisory firm Volvér showcase the significance of science and risk in devising a manufacturing facility.
How can cannabis producers bring a high quality, reliable product onto the market? Morten Allesoe, Principal Consultant and Christian Carlsen, Venture Partner at independent consulting firm NNE, tell MCN about the importance of factoring in scientific evidence and risk awareness.
“We have previously discussed various steps in the journey from having an idea as an investor or entrepreneur, to creating and delivering the product to the patient and the processes that take place within the body once the patient consumes the product – but we are still missing some of the steps between the initial idea and the launch of an operational facility,” explains Business Director Christian Carlsen.
“We advise our customers to take the science- and risk-based approach with a heavy patient focus, but this is a factor that migrates into the broader strategy on how to qualify the facility and the equipment,” says Principal Consultant Morten Allesoe.
How can a focus on science and risk help solve the challenges faced by medical cannabis producers in an emerging market?
Pharmaceutical engineering and consulting company NNE has provided advice and support to an increasing number of medical cannabis clients in recent years. As the nascent medical cannabis industry has grown and developed, both in Europe and elsewhere, Carlsen has identified some key differences between the field of medical cannabis and the traditional pharmaceutical sector.
“Medical cannabis is a relatively young and very American industry,” he explains. “A lot of medical cannabis operators and stakeholders have very little experience or prior knowledge of the pharmaceutical industry.” This is where NNE’s extensive background in the pharmaceutical sector comes in.
“We often hear clients say that they want to produce a special, high quality product: we normally tell these clients that ‘quality’ is not a specific category for pharmaceutical products – quality is one of the main pillars of the industry; you cannot deliberately produce a low quality pharmaceutical product, because then the product will never be released,” says Carlsen.
“Product quality standards are very well-defined in pharmacopoeias,” Allesoe adds. “The regulatory requirements are laid out in monographs; and there are well-defined monographs for the cannabis flower – you could do more from a quality standpoint, but it’s not required from a patient or regulatory perspective; and it is not something we advise customers to do. There is one key demand on product quality: it must meet regulatory requirements.”
In an emerging market with a high sense of urgency, time to market is key. “In general, we have found that everybody within this industry has a high sense of urgency and they want to have everything out on the market yesterday,” Carlsen comments. Medical cannabis producers wishing to reduce the implementation time of their site may do so by either increasing costs or reducing the scope of their facility – raising costs necessitates the allocation of increased resources, which may not be a feasible solution for investor-backed clients with a limited financial base.
Allesoe explains: “People have to be aware that when they ask for the same quality, but they want it to be done faster, there are essentially two ways to solve this (Fig. 1). You can either bring the cost up, by adding or allocating more resources – and in this context, this is not something that we advise our clients to do. Another way to solve it is to decrease the scope, by saying: if we want to do this in a shorter time, we cannot do the same things; we have to work smarter, not harder – this really works. The Food and Drug Administration (FDA) guidance on process validation supports this method, which is a science- and risk-based approach, emphasising the need to focus on the patient impact when setting critical requirements. The methodology is covered by the ASTM E2500 standard: ‘Standard Guide for Specification, Design, and Verification of Pharmaceutical and Biopharmaceutical Manufacturing Systems and Equipment’. This approach means that a lot of the requirements which used to be critical in traditional qualification protocols become non-critical, meaning that you can ease your level of testing and rely more heavily on the supplied documentation. You will still have to test everything which is critical to the patient, defined as having an impact on the so-called Critical Quality Attributes, but the testing process is heavily reduced in scope.”
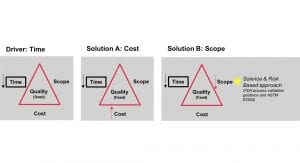
NNE has seen the number of critical requirements for individual facilities fall by more than half by implementing this new approach to qualification and validation, while the documentation load is reduced by around 90%.
Allesoe highlights the implications of this: “Especially in the fact that you have significantly fewer critical requirements, that means that you have to maintain these to a much smaller extent, because as soon as you deem something to be critical, it means that you have to monitor it throughout commercial operation as part of continued process verification following product launch. It is a huge burden to have too many of these – especially if they are not relevant to the patient experience – but once a critical requirement has been implemented it is difficult to remove, because it’s included in the documentation submitted to the licensing authorities.”
Even where a facility has begun its validation process in the traditional manner, it may still be possible to reduce some of these critical requirements at a later stage, Allesoe clarifies.
“You would have to determine the workload involved in revisiting or changing the risk assessment, because risk assessments are typically quite extensive and complex: it is possible to reduce the burden they incur, but you need to put a lot of work into it and the documentation must then be reapproved by the licensing authorities. Within the traditional approach, the risk protocol often entails conducting many different risk assessments; and you have to go through all of them to ensure that your removal of these requirements does not impact the validated state – the ultimate goal is to have a validated process for manufacturing your medical product.”
That is why NNE advises its clients to embed multiple risk assessments into a single holistic quality risk assessment covering the entire process and with direct linkage to the Critical Quality Attributes, defined as the quality metrics relevant to the patient (Fig. 2).
The science- and risk-based approach
The science- and risk-based approach to converting an initial concept into a certified operational facility is relatively cost efficient, because it has fewer critical requirements than traditional validation methods. In addition to the economic benefits of this approach, it streamlines and accelerates the process of bringing a product to market – however, Allesoe emphasises that both the science-based and traditional approaches are broadly effective; and that they both have the same final result: a high quality, marketable finished product.
“Many roads lead to Rome,” he says. “This is just an approach that is leaner, smarter and easier to maintain during the commercial operation; all made possible to focus solely on the critical-to-patient aspects of the product and process.”
The traditional approach to facility validation, sometimes referred to as the V model due to the shape of its integral process flowchart, is based on guidance drawn up by the European Medicines Agency. Relatively speaking, the different steps of the process are distinctly disconnected from each other under this approach: different specifications must be implemented discretely for the user requirement, functional and design aspects of the process; then once the equipment has been received, installation qualification (IQ), operation qualification (OQ) and performance qualification (PQ) must each be separately gauged.
Allesoe outlines the flaws in this method: “This process results in a lot of documentation, but it is often not linked to risk assessments rooted in the critical quality attributes which are necessary to the patient experience. It entails a lot of critical requirements, a lot of paperwork, a lot of repetitions of what the supplier has already done; and you end up testing things again which the supplier has already tested. There’s a lot of room for optimisation.”
By contrast, the science- and risk-based approach, which is based on standards set by the American Society for Testing and Materials (ASTM) and is referenced in the FDA’s official guidance for industry and process validation, considers the validation process in much the same way as a manufacturing system: rather than viewing each stage as a single entity, the entire process is assessed holistically. This highlights the few critical vectors of the process where further testing is necessary, as the other aspects will already have been subjected to extensive testing by suppliers and vendors; thereby significantly reducing the need for extraneous testing protocols.
Some producers experience a level of confusion over the authorisation of each approach, assuming that facilities producing for the European market must comply solely to the EMA’s guidelines. This is not the case, says Allesoe. “Some people think that they are not allowed to rely on FDA process validation guidance, but the EMA and FDA have a mutual recognition agreement: they are allowed to rely on each other’s inspection metrics and process guidance; there is a harmonisation between the two. It is fully acceptable for a company that supplies to the European market to build and qualify according to the science- and risk-based approach as outlined in FDA’s Process Validation guidance and ASTM E2500. Sometimes we do still have to convey this message to the authorities on behalf of our clients at the inspection stage, and they are fully receptive.”
Quality risk management, GEP and supplier collaboration
Quality risk management is a key aspect at each stage of the science- and risk-based validation process, Allesoe emphasises: “We have to consider the critical quality attributes of the product: what is relevant to the patient, what can harm the patient, what can ensure that the product will be effective. This is what you build your risk assessment on; it impacts the level of testing in all phases of production. In addition to the initial quality risk assessment, you do later iterations as you learn more and are able to go into more detail. Then you keep refining your risk assessment until you end up with the control strategy that binds all of this together.
Good Engineering Practice (GEP), another significant aspect underpinning the validation process, is driven primarily by the subject matter expert.
“The role of the subject matter expert is much more emphasised in the science- and risk-based approach than the traditional approach, where the quality assurance (QA) department has a huge role throughout the entire scheme and especially throughout qualification,” says Allesoe. “Here we acknowledge that the people who really know the process are also the ones who are most capable of setting the right requirements. Supplier collaboration is also very key in this approach: we have seen many clients who tend to repeat what the supplier has already fully documented in terms of compliance, which is a huge waste of work because often the supplier understands the equipment better. It should be acceptable – and it is, according to this approach – to rely more on documentation signed by suppliers, so now our clients are allowed to rely on supplier documentation – following a vendor assessment, of course.”
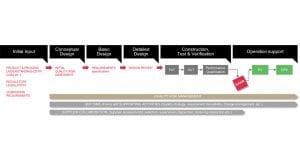
Step 1: quality risk assessment and facility design
At the earliest stage of implementation, before the design phase, initial inputs are captured. These may include process understanding gained from, for instance, previous development studies, critical and non-critical corporate requirements; as well as regulatory standards that must be taken into account. Once the initial inputs have been gathered, the design of a planned facility can begin to be conceptualised. At this stage, Carlsen notes, a simple risk assessment becomes necessary. “We do a very simple, two-dimensional layout of the facility, some rough budgeting around the project and we draw up a timeline,” he says.
“This is the stage where we try to put the idea down to paper, in order to determine whether the idea in its current form will work or not – by looking into the cost and revenue projections, we can qualify how feasible the concept is; and from there, we can decide whether we want to go to the next stage. This takes us a step deeper into the documentation: the drawings become more detailed; the quality risk assessments become more in-depth; the budget becomes more comprehensive. Then, once you have a fully designed, executable project, you can hand it over to a construction company to begin physically establishing the facility.”
Step 2: construction, testing and verification
Once a facility has been designed, a producer can begin drawing up user requirement specifications in preparation for purchasing the necessary equipment. New equipment is subject to both a factory acceptance test (FAT) at the supplier’s site and a site acceptance test (SAT) once it has been installed at the user’s facility: under the traditional approach, the SAT is conducted by the supplier and then repeated by the manufacturer with several elements contained in the IQ/OQ testing.
With the science- and risk-based approach, however, SATs can be performed in collaboration between the supplier and the manufacturer: Installation Qualification (IQ) and Operational Qualification (OQ) documentation provided by the supplier can be taken into account in the testing process; and if the manufacturer still feels additional testing needs to be conducted they can do so. Performance Qualification (PQ) for a new facility involves worst-case parameter testing of the production process, in order to determine that the process is functional before the equipment can be released: the results of FAT/SAT and PQ testing are then documented in a system acceptance release report (SARR).
Step 3: final approval and operational support
“When we have finalised the construction, testing and verification, then we have a finalised facility,” Carlsen says. “This means that the facility is ready for Good Manufacturing Practice (GMP) inspection, because we have built, tested and verified the facility against the documentation that we have created before we started the construction. Now that the facility is ready for inspection, we can invite the authorities to inspect the facility in order to get a GMP approval; then when it is GMP approved, it is ready for GMP production.”
Allesoe adds: “Once a facility receives GMP approval, it is possible to initiate Process Validation activities related to one or several products: this is where you verify and validate the control strategy for the product as anchored in the quality risk assessment. As this is a life cycle approach, there is an additional protocol following product launch called ongoing process verification (OPV), which means that in principle, the PV never stops. It continues even after the product has been launched – when your product is on the market, you still keep refining your control strategy, because you gain new knowledge as you accumulate new data.
“This is the whole life cycle. It starts from the initial input – from people having an idea, sitting around the table, saying: we want to make medical cannabis products for this patient target group. Then they start building their design, setting specifications for construction and for the process equipment; and then in the end, they use everything that they have built to make a product. I think it’s beautiful: it binds together the design, construction, testing and verification, which used to be conducted exclusively by engineers, with the product itself and the patient aspect, which is the focus of the pharmacists. This strategy is unique, and it makes the process so much more transparent.”
Uncertainty and risk in an emerging industry
The additional speed and transparency offered by the science- and risk-based approach are particularly significant in the cannabis sector, as an emerging industry with limited in-depth expertise among its operators. Carlsen notes: “We often see people who just want to build a cannabis facility which will deliver products very fast, but they don’t know the patient target group and they don’t know what products the market needs. While there are a lot of opportunities in this industry, there is also a lot of uncertainty; so we have to be somehow flexible around the product and patient side, in order to create different products for different patient groups – we have to adapt to meet new requirements in order to match the behaviour of the emerging industry.”
“We challenge clients quite a lot,” adds Allesoe. “We meet with clients who have an idea, but they just want to start building right away – they have so many acres, they want to cultivate, they want to extract cannabis resin and they want to make oils, but that’s it. As a starting point, we insist that they devise a quality target product profile (QTPP) and an early stage qualitative risk assessment which helps them get to know their product and their target base from the beginning. We have to trigger reflection around the patient and the product from day one: this is good practice in consulting. We want our clients to be a success; we want to make sure that we don’t set a process in motion where we don’t really know the end goal, because it would be a failure for the clients, but also for us as a consulting company. This is the take-home message: we advise our clients to make these initial considerations on what they want to produce, because then we can apply the right strategy.”
Christian Carlsen
Venture Partner
Morten Allesoe, PhD Pharm
Principal Consultant
czca@nne.com
cc@volverventures.com
Tweet @NNEglobal
www.nne.com
This article is for issue 4 of Medical Cannabis Network. Click here to get your free subscription today.






